スフィンゴ脂質恒常性維持のためのセラミド分解経路
2023.10.28
木原章雄*
*北海道大学大学院薬学研究院
1.はじめに
生体分子は合成と分解のバランスによって恒常性が保たれている。そのため,合成だけでなく,分解に異常が生じた場合も疾患に結びつくことが多い。スフィンゴ脂質に関しても分解経路の先天性疾患としてスフィンゴリピドーシスがよく知られている。それぞれの生体分子には驚くほど巧妙かつ無駄のない分解系が備わっており,単に分解するというだけでなく,他の生体分子の前駆体あるいはエネルギー源として利用できる分子へと変換する。このことはセラミドについても当てはまり,セラミドの長鎖塩基部分は長鎖アルデヒドとホスホエタノールアミンへと分解された後,様々な脂質の生合成あるいはエネルギー産生(β酸化)に利用される。長鎖塩基から脂肪族アルデヒドを経てアシルCoAへと至る分解経路の詳細は長年不明であったが,近年筆者らのグループが解明に成功した。筆者らが同定した長鎖塩基代謝遺伝子には先天性疾患の原因遺伝子が含まれていた。本稿では,これらの知見を含めたセラミド/長鎖塩基分解経路の詳細,セラミド代謝異常と疾患との関連について紹介する。
2.セラミドの構造
スフィンゴ脂質骨格セラミドは長鎖塩基と脂肪酸がアミド結合した構造をもつ(図1A)。長鎖塩基の化学構造には生物間で違いがあるが,哺乳類にはスフィンゴシン,ジヒドロスフィンゴシン(スフィンガニン),フィトスフィンゴシン,6-ヒドロキシスフィンゴシン,4,14-スフィンガジエンの少なくとも5種類が存在する1, 2)(図1B)。長鎖塩基は共通してC1位とC3位に水酸基,C2位にアミノ基をもつ。ジヒドロスフィンゴシンはこの3つの官能基しかもたない最も単純な長鎖塩基である。哺乳類の長鎖塩基の炭素鎖長はC18が最も多く,ジヒドロスフィンゴシンはd18:0と表記される。dは2つの水酸基,18は炭素鎖長,0は二重結合の数を表す。哺乳類に最も多い長鎖塩基はスフィンゴシン(4E-d18:1)であり,C4位とC5位の間にトランス二重結合をもつ。4,14-スフィンガジエン(4E,14Z-d18:2)はスフィンゴシン骨格に対してC14位とC15位間にシス二重結合をもつ3)。これら3種の長鎖塩基は全身に存在する。一方,C4位に水酸基をもつフィトスフィンゴシン(t18:0)は表皮,小腸,腎臓などの限られた組織に存在する2)。6-ヒドロキシスフィンゴシン(t18:1)はスフィンゴシン骨格に対してC6位に水酸基をもち,表皮特異的に存在する2, 4)。
脂肪酸は様々な脂質の構成成分として利用されるが,スフィンゴ脂質中の脂肪酸には他の脂質中の脂肪酸にはない特徴がある。脂肪酸は炭素鎖長の違いから,短鎖(C2からC4),中鎖(C5からC10),長鎖(C11からC20),極長鎖(C21以上)に分類される。また,二重結合の数から飽和,一価不飽和,多価不飽和に分類される。グリセロリン脂質中の脂肪酸の殆どは長鎖の飽和,一価不飽和,多価不飽和脂肪酸(C16:0,C16:1,C18:0,C18:1,C18:2,C20:4)である5)。一方,スフィンゴ脂質中の脂肪酸は殆どの組織ではC16:0,C18:0,C20:0,C22:0,C24:0,C24:1,C24:2が主要である6, 7)。ただし,これらの割合は組織ごとに異なり,神経細胞ではC18:0脂肪酸(C18:0-COOH)を含むスフィンゴ脂質が多く,肝臓ではC24:0-,C24:1-COOHを含むものが多い。C16:0-,C18:0-COOHがグリセロリン脂質とスフィンゴ脂質中に共通に見られるのに対して,C20:0-,C22:0-,C24:0-,C24:1-,C24:2-COOHはほぼスフィンゴ脂質に特異的に存在する。例外的に精巣,精子にはC28からC34の4価から6価の多価不飽和極長鎖脂肪酸含有スフィンゴ脂質6, 8, 9),表皮にはC26からC36の飽和/一価不飽和極長鎖脂肪酸含有スフィンゴ脂質が存在する4, 10)。
スフィンゴ脂質の脂肪酸の多くは非水酸化型であるが,2位(α位)またはω位に水酸基をもつものも存在する(図1C)。これらの水酸化脂肪酸はグリセロリン脂質中には存在しない。2-水酸化(2-OH)脂肪酸を含むスフィンゴ脂質は脳ミエリン,表皮,小腸などの限られた組織に存在する11)。特に脳ミエリンに存在するガラクトシルセラミドの約2/3は2-OH脂肪酸を含有しており,ミエリンの形成と維持に重要である12)。脂肪酸2-水酸化酵素FA2Hはこの2-OH化を触媒する酵素であり,FA2H遺伝子に変異が生じると遺伝性痙性対麻痺(SPG35)を引き起こす13, 14)。表皮にはC30からC36のω-水酸化(ω-OH)脂肪酸を含むセラミドが存在している4)。ただし,ω位水酸基が遊離の状態で存在するセラミドは少なく,殆どがリノール酸とエステル結合をしている。このような構造をもつセラミドはω-O-アシルセラミド(アシルセラミド)と呼ばれる2, 15)。ω-OH位の水酸化はシトクロームP450ファミリーに属するCYP4F22によって触媒される16)。アシルセラミドは皮膚バリア機能に重要であり,CYP4F22を含めてアシルセラミド産生に関わる遺伝子の変異は魚鱗癬と呼ばれる皮膚角化症を引き起こす2, 17-19)。
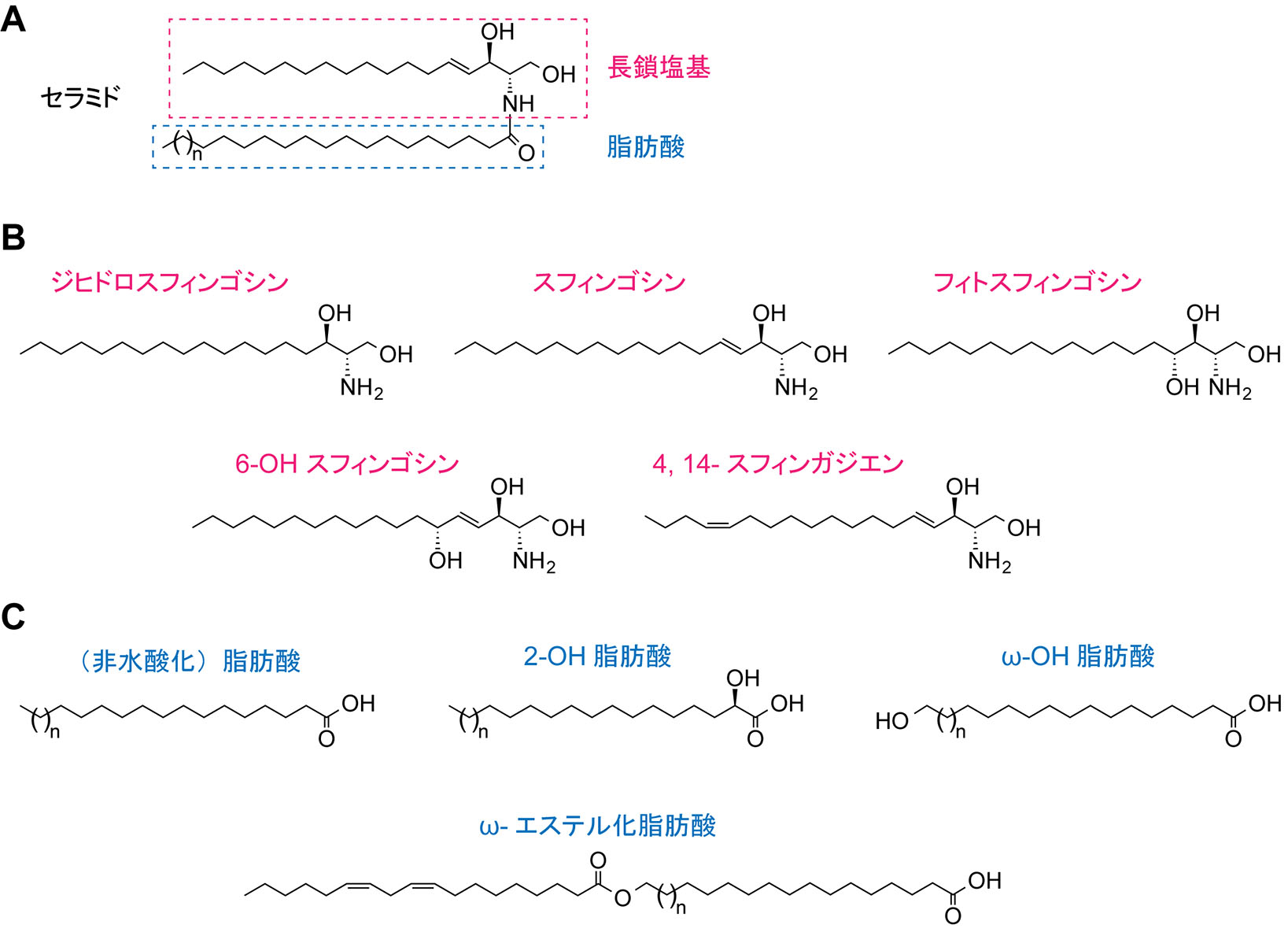
図1 セラミドの構造
3.長鎖塩基分解経路の概要
複合スフィンゴ脂質(スフィンゴミエリン及びスフィンゴ糖脂質)は主にリソソーム中で脂肪酸と長鎖塩基にまで分解される。これらの分解に関わる遺伝子の変異はスフィンゴリピドーシスと呼ばれる一群の代謝異常症を引き起こす20)。例えば,セラミドを分解するセラミダーゼ遺伝子ASAH1の変異はFarber病を引き起こす21)。Farber病は関節腫脹変形,皮下結節,喉頭障害の症状を伴う。セラミダーゼには至適pHの異なる3つのタイプ(酸性,中性,アルカリ性セラミダーゼ)が存在するが,ASAH1は酸性セラミダーゼに分類される22)。中性セラミダーゼにはASAH2,アルカリ性セラミダーゼにはACER1,ACER2,ACER3が存在する。
セラミドという用語は広義ではどのタイプの長鎖塩基を含むものも指す。一方,狭義では長鎖塩基がスフィンゴシンであるものだけを指す。後者の場合,ジヒドロスフィンゴシンをもつものをジヒドロセラミド,フィトスフィンゴシンをもつものをフィトセラミドと呼ぶ。セラミダーゼによって生じた長鎖塩基はスフィンゴ脂質合成に再利用されるか,分解経路(異化経路)によってアシルCoAへ変換される。再利用経路(サルベージ経路)では,セラミド合成酵素がセラミド(広義)を産生する。哺乳類には6種のセラミド合成酵素(CERS1からCERS6)が存在し,それぞれが異なった鎖長のアシルCoAを基質とする2)。長鎖塩基分解経路によって生じたアシルCoAは脂質の前駆体(主にグリセロリン脂質)として使用されるか,β酸化によってエネルギー産生に利用される。長鎖塩基のどのくらいの割合がサルベージ経路あるいは分解経路に供されるかは,細胞の種類や生育環境によって異なる。筆者らが以前に放射標識ジヒドロスフィンゴシンを用いて細胞をラベルした実験では,ジヒドロスフィンゴシンの6から8割がサルベージ経路によってスフィンゴ脂質へ,2から4割が分解経路によってグリセロ脂質へ代謝された23)。この実験では分解経路で生じたアシルCoAのどのくらいの割合がβ酸化によってCO2へ代謝されたか検討できていないが,ラットに静脈注射した放射標識ジヒドロスフィンゴシンの放射活性の約25%が10時間後にCO2へ取り込まれたという報告がある24)。長鎖塩基の代謝によって生じたアシルCoAとde novo合成経路によって生じたアシルCoAは区別なく代謝されるようである。
長鎖塩基の分解経路において,長鎖塩基はリン酸化,開裂,酸化,CoA付加という4つの基本反応によってアシルCoAへ変換される2, 25)(図2)。最も単純な長鎖塩基であるジヒドロスフィンゴシン(d18:0)は,これらの4反応によってパルミトイルCoA(C16:0-CoA)へ変換される。スフィンゴシン(d18:1)はジヒドロスフィンゴシンと同様にC16:0-CoAへ代謝されるが,そのためには基本4反応に飽和化のステップが加わる26)(図3)。フィトスフィンゴシン(t18:0)は水酸基をC4にもつため,基本4反応によって2-OH C16:0-CoAへ変換される(図4)。2-OH脂肪酸はスフィンゴ脂質中にのみ見られ,グリセロリン脂質合成には使用されないため,2-OH C16:0-CoAの状態では利用が限られる。このことを回避するため,生体はα酸化という1炭素の減少を伴った水酸基の除去法を備えている。これにより,フィトスフィンゴシンはペンタデカノイルCoA(C15:0-CoA)となり,C16:0-CoAと同様,脂質合成(主にグリセロリン脂質)あるいはβ酸化に利用される。6-ヒドロキシスフィンゴシンの代謝経路の詳細は不明である。6-ヒドロキシスフィンゴシンは表皮の最外層である角質層に多く存在し,皮膚バリア機能において重要な役割を果たしていると考えられている。角質層の細胞(角質細胞)は死細胞であり,垢となって除去されることから,6-ヒドロキシスフィンゴシンは代謝を受けない可能性がある。4,14-スフィンガジエンの代謝経路はスフィンゴシンと同様である。すなわち,基本4反応後にC4位のトランス二重結合が飽和化され,C16:1-CoAとなる27)。もともとC14位にあったシス二重結合は飽和化されない。
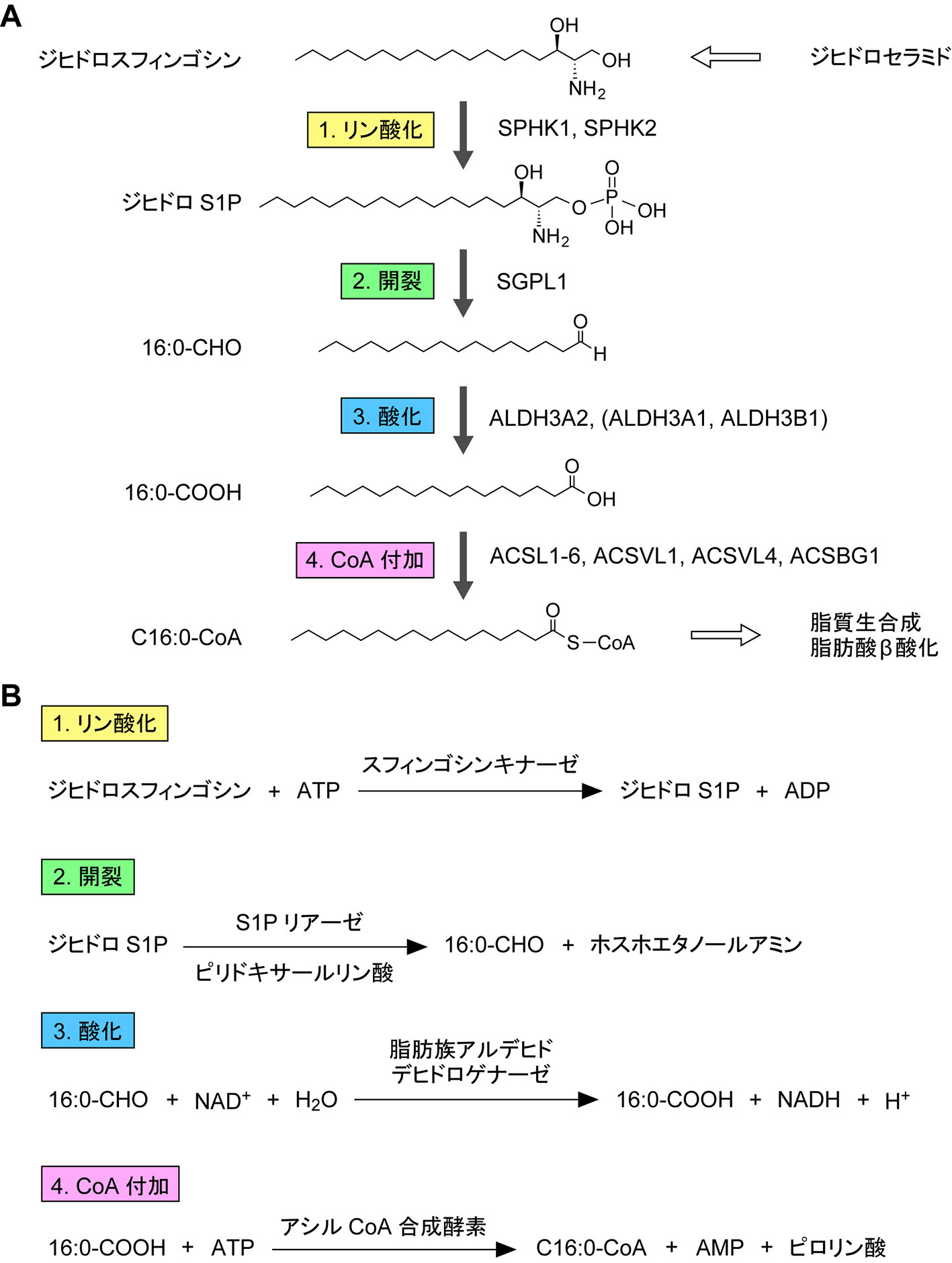
図2 ジヒドロスフィンゴシン分解経路
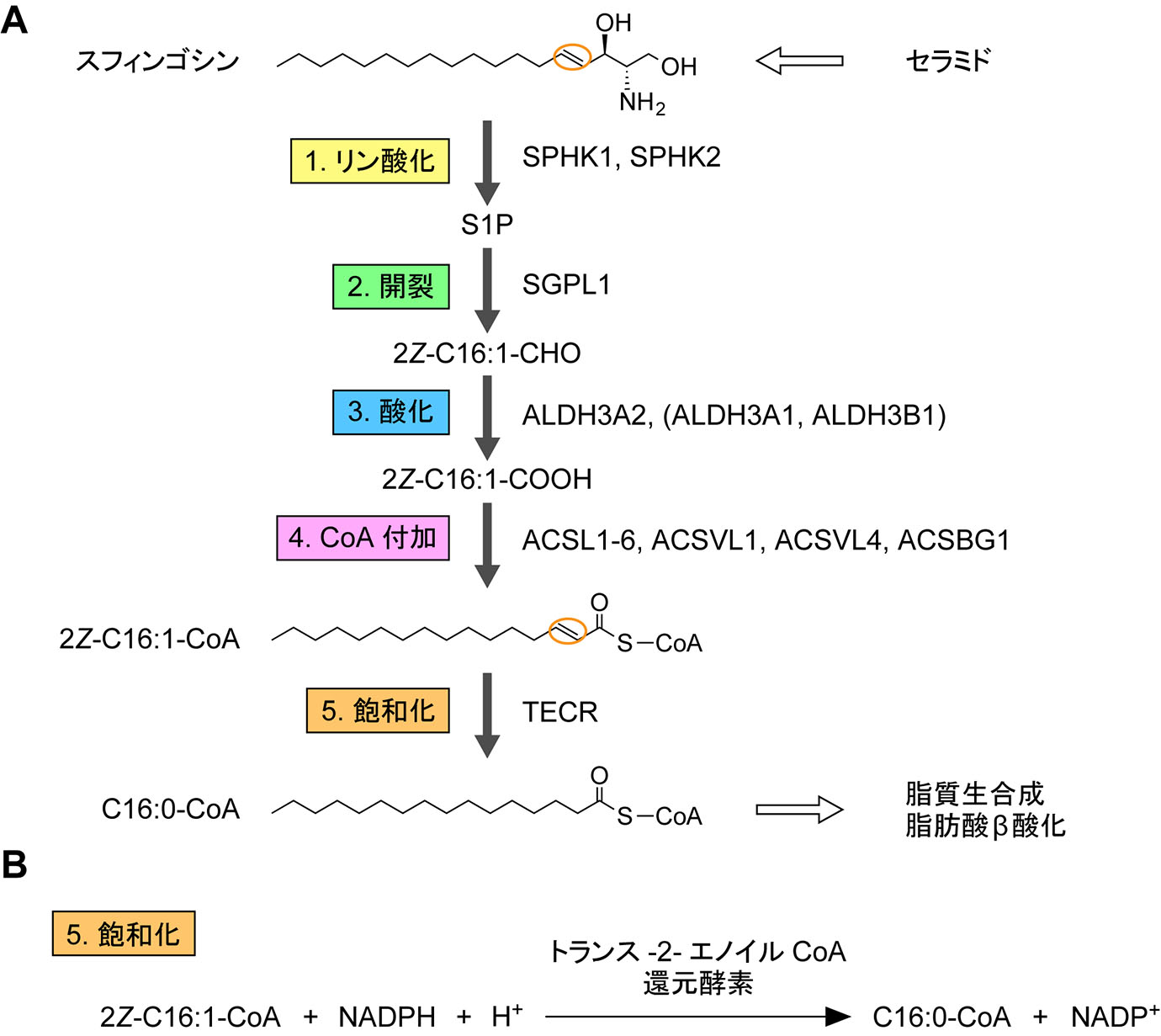
図3 スフィンゴシン分解経路
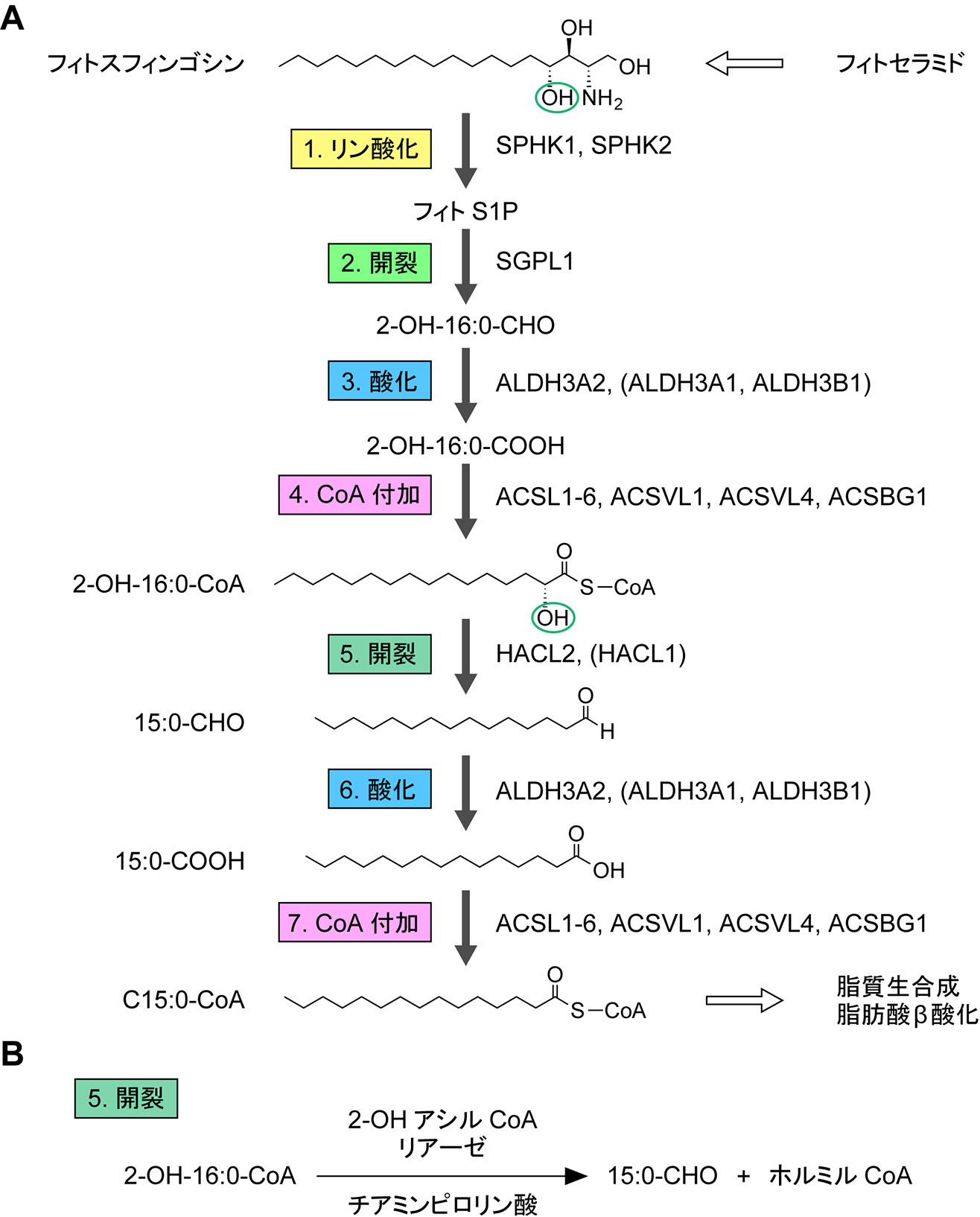
図4 フィトスフィンゴシン分解経路
4.長鎖塩基分解経路における反応
長鎖塩基代謝の第一段階は長鎖塩基のリン酸化であり,スフィンゴシンキナーゼによって触媒される。哺乳類にはSPHK1とSPHK2の2種類のアイソザイムが存在する28, 29)。ジヒドロスフィンゴシン,スフィンゴシン,フィトスフィンゴシンはスフィンゴシンキナーゼによってそれぞれジヒドロスフィンゴシン1-リン酸(ジヒドロS1P),スフィンゴシン1-リン酸(S1P),フィトスフィンゴシン1-リン酸(フィトS1P)へ変換される(図2–4)。これらの長鎖塩基1-リン酸は細胞内で産生された後,殆どは引き続く開裂反応によって速やかに分解される。一方で,これらの長鎖塩基1-リン酸,特にS1Pは脂質メディエーターとしても機能する30)。S1Pは内皮細胞(血管内皮細胞,リンパ管内皮細胞)あるいは血球(赤血球,血小板)で産生された後,一部が細胞外へ放出され,細胞表面に発現している受容体(S1P受容体S1P R1からS1PR5)に結合して様々な細胞応答を引き起こす30, 31)。
長鎖塩基1-リン酸は脱リン酸化によって再度長鎖塩基に戻されるか,不可逆的にC2位とC3位間の開裂反応を受けて長鎖アルデヒドとホスホエタノールアミンになる(図2B)。前者はS1Pホスファターゼ(SGPP1,SGPP2),後者はS1Pリアーゼ(SGPL1)によって触媒される32-34)。これらの酵素はいずれも小胞体に局在する34, 35)。SGPL1はピリドキサールリン酸依存性酵素である。SGPL1による開裂反応によってジヒドロS1P,S1P,フィトS1Pからそれぞれヘキサデカナール(パルミトアルデヒド;C16:0-CHO),トランス-2-ヘキサデセナール(2Z-C16:1-CHO),2-ヒドロキシヘキサデカナール(2-OH C16:0-CHO)が産生される(図2–4)。SGPL1のもう1つの生成物であるホスホエタノールアミンはCDP-エタノールアミンへ変換後,Kennedy経路によってグリセロリン脂質の1つであるホスファチジルエタノールアミンへ代謝される36, 37)。
S1Pリアーゼによる開裂反応は長鎖塩基代謝の最初の不可逆反応であり,SGPL1が唯一のS1Pリアーゼ遺伝子であることから,この遺伝子の欠損によって長鎖塩基代謝が完全に停止する。従って,Sgpl1ノックアウト(KO)マウスの表現型からスフィンゴ脂質恒常性の破綻の影響が読み取れる。Sgpl1 KOマウスは体が小さく,短命であり,生後20日から50日の間で死亡する38)。組織学的な解析では,肺,心臓,尿路,骨において異常が見つかっている39)。肝臓における脂質解析では,SGPL1の基質であるS1Pだけでなく,セラミドやスフィンゴミエリン量も上昇していた38)。これは蓄積したS1PがS1Pホスファターゼによる脱リン酸化を受けて長鎖塩基へ変換後,セラミド,さらにスフィンゴミエリンへと代謝されたためである。このような広範にわたる組織への影響はスフィンゴ脂質恒常性維持の重要性を物語っている。また,SGPL1遺伝子変異はヒトではステロイド抵抗性ネフローゼ症候群を引き起こす。この疾患はネフローゼ以外に魚鱗癬,副腎不全,免疫不全,神経障害,筋萎縮,甲状腺機能低下,停留睾丸症を伴う40-42)。
S1Pリアーゼによって生じた長鎖アルデヒドは,アルデヒドデヒドロゲナーゼ(ALDH)による酸化を受け,長鎖脂肪酸となる。その結果,ジヒドロスフィンゴシン,スフィンゴシン,フィトスフィンゴシンからそれぞれパルミチン酸(C16:0-COOH),トランス-2-ヘキサデセン酸(2Z-C16:1-COOH),2-OHパルミチン酸(2-OH C16:0-COOH)が産生される(図2–4)。この反応における酸化剤はNAD+である(図2B)。ヒト及びマウスにはALDHがそれぞれ19種と21種存在する43, 44)。これらのうち,長鎖アルデヒドに高い活性を示すALDHはALDH3サブファミリーである45, 46)。ヒトにはALDH3サブファミリーが3種(ALDH3A1,ALDH3A2,ALDH3B1),マウスには5種(Aldh3a1,Aldh3a2,Aldh3b1,Aldh3b2,Aldh3b3)存在する。ヒトのALDH3B2は偽遺伝子であり,機能を失っている。また,ヒトにはALDH3B3が存在しない。ALDH3サブファミリーメンバーの細胞内局在は異なっており,ALDH3A1/Aldh3a1がサイトゾル,ALDH3A2/Aldh3a2が小胞体,ALDH3B1/Aldh3b1とAldh3b3が細胞膜,Aldh3b2が脂肪滴に局在する46, 47)。S1Pリアーゼによって長鎖アルデヒドは小胞体で産生される35)。そのため,ALDH3ファミリーメンバーの中で長鎖塩基代謝経路で主要に働くのは小胞体に局在するALDH3A2/Aldh3a2である。Aldh3a2が欠損したCHO-K1細胞中ではスフィンゴシンからグリセロリン脂質への代謝が野生型細胞中に比べて2割程度まで低下する48)。
ALDH3A2はSjögren-Larsson症候群の原因遺伝子である。この疾患は皮膚神経疾患であり,魚鱗癬,痙性対麻痺,精神遅滞を特徴とする49)。上述の通り,ALDH3A2遺伝子が完全に機能を失っても他のALDH3サブファミリーメンバーにより,部分的に長鎖塩基代謝は進行する。そのため,SGPL1遺伝子変異により引き起こされるステロイド抵抗性ネフローゼ症候群とは異なり,Sjögren-Larsson症候群の発症原因はスフィンゴ脂質恒常性の異常が主な原因ではないと思われる。実際,Aldh3a2 KOマウス由来ケラチノサイトでは,長鎖塩基の代謝は低下しているが,セラミド量は殆ど変化していない48)。むしろSjögren-Larsson症候群の病態はALDH3A2遺伝子変異によって蓄積するアルデヒドが引き起こすと考えられている50)。アルデヒドのカルボニル炭素と酸素の電気陰性度の違いは電荷の偏りを生じさせる。そのため,カルボニル炭素は求核攻撃を受けやすく,アルデヒドはリシンなどのもつ第一級アミンとシッフ塩基を形成しやすい。さらに,アルデヒドの中でもスフィンゴシン由来の2Z-C16:1-CHOはα,β不飽和アルデヒドに分類され,特に反応性が高い51)。α,β不飽和アルデヒドは第一級アミンだけでなく,ヒスチジンやシステイン側鎖がもつ一般的な求核基ともMichael付加反応を行う。その中でも,2Z-C16:1-CHOがヒスチジンと安定な共有結合体を形成することがin vitroの実験から示されている52)。筆者らは,Aldh3a2 KOマウスの脳においてミエリンの形成/維持に重要な2-OHガラクトシルセラミドの量が減少していることを見出した51)。この原因として,Aldh3a2 KOマウス中で蓄積したアルデヒド(特にスフィンゴシンの分解産物である2Z-C16:1-CHO)が,脂肪酸2-水酸化酵素FA2Hを攻撃したことが考えられる。また,Sjögren-Larsson症候群モデルマウス(Aldh3a2 Aldh3b2二重KOマウス)は魚鱗癬様の皮膚バリア異常を示し,その原因は皮膚バリア機能において重要な働きをするアシルセラミドの産生低下であった53)。このアシルセラミドの産生低下はSjögren-Larsson症候群患者の角質層においても観察された54)。このことから,2Z-C16:1-CHOは表皮においてアシルセラミド産生に関わる酵素も攻撃し,活性を低下させていると考えられる。
脂肪酸はそのままでは代謝されず,アシルCoAへ変換されて活性化される必要がある。ALDH3A2によって産生された長鎖脂肪酸も同様であり,ジヒドロスフィンゴシン,スフィンゴシン,フィトスフィンゴシンの代謝経路からそれぞれC16:0-CoA,トランス-2-ヘキサデセノイルCoA(2Z-C16:1-CoA),2-OH C16:0-CoAが産生される(図2-4)。脂肪酸にCoAを付加する反応はアシルCoA合成酵素(ACS)によって触媒され,合成のエネルギーはATPのAMPとピロリン酸への加水分解である(図2B)。ヒトには26種類のACSが存在し,基質特異性と配列の相同性から6つのサブファミリーACSS(ACS short-chain),ACSM(ACS medium-chain),ACSL(ACS long-chain),ACSVL(ACS very long-chain),ACSBG(ACS bubblegum),ACSF(ACS family)に分類される55)。このうち,長鎖塩基代謝において働くのは,長鎖脂肪酸に高い活性を示すACSLファミリーメンバー(ACSL1,ACSL3,ACSL4,ACSL5,ACSL6)とACSVL1,ACSVL4,ACSBG1である26, 56)。これらは重複した活性を示すので,特定の1つのACS遺伝子に変異が入っても長鎖塩基分解経路は殆ど影響を受けない。
5.スフィンゴシン分解経路における飽和化反応
スフィンゴシンは上記の4つの基本反応によって2Z-C16:1-CoAへ変換される(図3A)。グリセロリン脂質には2Z-C16:1-COOHをもつものは殆ど存在せず,2Z-C16:1-CoAは飽和化してC16:0-CoAとなってからグリセロリン脂質に取り込まれる。ただし,2Z-C16:1-CoAはβ酸化の中間体となり得るため,β酸化のためには飽和化される必要はないのかもしれない。2Z-C16:1-CoAを飽和化する酵素はトランス-2-エノイルCoA還元酵素であり,哺乳類ではTECRがコードしている23)。TECRによる飽和化(還元)反応ではNADPHが還元剤として使用される(図3B)。TECRはもともと脂肪酸伸長サイクルにおいて機能する遺伝子として同定されたものである57)。脂肪酸の伸長はアシルCoAを基質として4つの反応(マロニルCoAから供給されたC2単位との縮合,還元,脱水,還元)を1サイクルとした脂肪酸伸長サイクルによって行われ,1サイクル毎にアシルCoAは炭素数を2つ増加する6, 9)。TECRは脂肪酸伸長サイクルの4段階目の還元反応を触媒する。従って,TECRは脂肪酸伸長サイクルと長鎖塩基分解経路の両方において機能する。脂肪酸伸長サイクルが産生する極長鎖脂肪酸は生存に必須であるため58),TECRの機能を大きく損なうような変異は胎生致死を引き起こすと予測される。ただし,TECRの弱い変異(p.Pro182Leu)による疾患は見つかっており,非症候性の精神遅滞を引き起こす59)。
6.フィトスフィンゴシン分解経路とα酸化
フィトスフィンゴシンは長鎖塩基代謝の基本4反応によって2-OH C16:0-CoAへ変換される(図4A)。産生された2-OH C16:0-CoAの一部はセラミド合成酵素の基質として用いられ,セラミドの脂肪酸部分へ取り込まれる60)。しかし,大部分の2-OH C16:0-CoAはα酸化を受けて非水酸化長鎖アルデヒド(ペンタデカナール;C15:0-CHO)となる。この反応は2-ヒドロキシアシルCoAリアーゼが触媒する(図4B)。これまで哺乳類の2-ヒドロキシアシルCoAリアーゼとしてHACL1が知られていた61)。しかし,HACL1はペルオキシソーム酵素であり,小胞体で行われる長鎖塩基代謝への寄与は小さい。筆者らは小胞体に局在する新規の2-ヒドロキシアシルCoAリアーゼHACL2を同定し,フィトスフィンゴシン代謝において主要な働きをしていることを明らかにした60)。HACL2はHACL1と配列の相同性を示し,共に補酵素としてチアミンピロリン酸を用いる。これまで脂肪酸α酸化はペルオキシソームで行われるというのが通説であったが,筆者らの結果は小胞体における脂肪酸α酸化の存在を初めて示したものである。
2-ヒドロキシアシルCoAリアーゼによって産生されたC15:0-CHOは,長鎖塩基の基本反応2によって産生されたC16アルデヒドと炭素数が1違うだけなので,同様の代謝を受ける。即ち,ALDH3A2によってC15:0-COOHへ酸化された後60),アシルCoA合成酵素によってペンタデカノイルCoA(C15:0-CoA)へ変換され,グリセロリン脂質などの脂質合成あるいはβ酸化に利用される。C15:0-CoAはβ酸化によって,6分子のアセチルCoA(C2:0-CoA)と1分子のプロピオニルCoA(C3:0-CoA)へ分解される。C3:0-CoAはプロピオニルCoAカルボキシラーゼによってD-メチルマロニルCoAと変換後,メチルマロニルCoAエピメラーゼによるL-メチルマロニルCoAへの変換を経て,補酵素B12依存的なメチルマロニルCoAムターゼによってスクシニルCoAとなり,クエン酸回路で代謝される62, 63)。
生体内の脂肪酸の殆どは偶数鎖であるが,奇数鎖脂肪酸も存在する。奇数鎖脂肪酸はFASによる生合成過程でアセチルACPの代わりにプロピオニルACPが使用されることによって産生されると以前は考えられていたが64, 65),その後の研究から主に2-OH脂肪酸のα酸化によって産生されるということが示唆されている66)。フィトスフィンゴシン分解経路から産生される2-OH C16:0-COOHは2-OH脂肪酸の供給源としては全体のごく一部であり,殆どは脂肪酸から脂肪酸2-水酸化酵素によって直接作られる。ミエリンには2-OH C24:0ガラクトシルセラミドが多く存在するが,その分解によって多くのC23:0-COOH(及び伸長したC25:0-COOHと不飽和化したC25:1-COOH)が産生される67)。それらから派生したC23/C25ガラクトシルセラミドは加齢と共に増加し,15歳児においてC23:0ガラクトシルセラミドはC24:0ガラクトシルセラミドの約1/3量に達する。表皮にも2-ヒドロキシ脂肪酸含有セラミドが多く存在するため,奇数鎖脂肪酸が多く,奇数鎖セラミドは偶数鎖セラミドの約半分程度存在する68)。筆者らは最近,脳と胃に多く存在する奇数鎖脂肪酸含有スフィンゴ脂質の奇数鎖脂肪酸の産生にHACL2が関与する2-OH脂肪酸のα酸化が主たる役割を果たすことを明らかにした69)。
7.おわりに
長鎖塩基がグリセロ脂質へと代謝されることは1960年代の後半に明らかにされた。また,長鎖塩基代謝の前半部に関わるスフィンゴシンキナーゼとS1Pリアーゼは1990年代の終わりに同定された。一方,長鎖塩基代謝の後半部の詳細と関与する遺伝子群は筆者らによって2010年代になってやっと明らかとなった。つまり,長鎖塩基分解経路の発見からその全容解明まで半世紀かかったわけである。今後は,これらの解明によって明らかになりつつある長鎖塩基代謝不全と疾患(Sjögren-Larsson症候群)の関係を明らかにし,治療法の開発に結びつけたい。
付記
本コンテンツは,セラミド研究会が編集した『セラミド研究の新展開~基礎から応用へ~』(発行日,2019年6月1日;発行者,株式会社・食品化学新聞社)に上梓した総説を一部内容改変して本研究会Webサイトに転載するものです.本コンテンツの著作権は食品化学新聞社に帰属しており,セラミド研究会は転載することの承認を食品化学新聞社から得ています.
本コンテンツは,クリエイティブ・コモンズ の定めたCC BY 4.0ライセンスの条件で掲載しており,著者と著作権所有者が明記され,かつ,日本セラミド研究会からの出版物である旨が引用されていることを条件として,他の会での使用,配布,または複製は許可されています.これらの条件に準拠していない使用,配布,または複製は許可されていません.
CC BY 4.0ライセンスの内容については以下URLを参照してください.
https://creativecommons.org/licenses/by/4.0/
図の説明
図1 セラミドの構造
A. セラミドの構造。B. 哺乳類における長鎖塩基の構造。C. セラミド中に存在する脂肪酸の構造。
図2 ジヒドロスフィンゴシン分解経路
A. ジヒドロスフィンゴシン分解経路における酵素反応と関与する酵素名。B. ジヒドロスフィンゴシン分解経路における各酵素反応の詳細。
図3 スフィンゴシン分解経路
A. スフィンゴシン分解経路における酵素反応と関与する酵素名。B. スフィンゴシン分解経路に固有の飽和化反応の詳細。
図4 フィトスフィンゴシン分解経路
A. フィトスフィンゴシン分解経路における酵素反応と関与する酵素名。B. フィトスフィンゴシン分解経路に固有の開裂反応の詳細。
引用文献
1) Pruett ST, et al.: Biodiversity of sphingoid bases (“sphingosines”) and related amino alcohols. J Lipid Res, 49, 1621-1639 (2008)
2) Kihara A: Synthesis and degradation pathways, functions, and pathology of ceramides and epidermal acylceramides. Prog Lipid Res, 63, 50-69 (2016)
3) Renkonen O, et al.: Structure of plasma sphingadienine. J Lipid Res, 10, 687-693 (1969)
4) t’Kindt R, et al.: Profiling and characterizing skin ceramides using reversed-phase liquid chromatography-quadrupole time-of-flight mass spectrometry. Anal Chem, 84, 403-411 (2012)
5) Yamashita A, et al.: Acyltransferases and transacylases involved in fatty acid remodeling of phospholipids and metabolism of bioactive lipids in mammalian cells. J Biochem, 122, 1-16 (1997)
6) Sassa T, et al.: Metabolism of very long-chain fatty acids: genes and pathophysiology. Biomol. Ther., 22, 83-92 (2014)
7) Edagawa M, et al.: Widespread tissue distribution and synthetic pathway of polyunsaturated C24:2 sphingolipids in mammals. Biochim Biophys Acta, 1863, 1441-1448 (2018)
8) Sandhoff R: Very long chain sphingolipids: tissue expression, function and synthesis. FEBS Lett, 584, 1907-1913 (2010)
9) Kihara A: Very long-chain fatty acids: elongation, physiology and related disorders. J Biochem, 152, 387-395 (2012)
10) Sassa T, et al.: Impaired epidermal permeability barrier in mice lacking Elovl1, the gene responsible for very-long-chain fatty acid production. Mol Cell Biol, 33, 2787-2796 (2013)
11) Hama H: Fatty acid 2-hydroxylation in mammalian sphingolipid biology. Biochim Biophys Acta, 1801, 405-414 (2010)
12) Zöller I, et al.: Absence of 2-hydroxylated sphingolipids is compatible with normal neural development but causes late-onset axon and myelin sheath degeneration. J Neurosci, 28, 9741-9754 (2008)
13) Alderson NL, et al.: The human FA2H gene encodes a fatty acid 2-hydroxylase. J Biol Chem, 279, 48562-48568 (2004)
14) Dick KJ, et al.: Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum Mutat, 31, E1251-E1260 (2010)
15) Uchida Y, et al.: Omega-O-acylceramide, a lipid essential for mammalian survival. J Dermatol Sci, 51, 77-87 (2008)
16) Ohno Y, et al.: Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation. Proc Natl Acad Sci USA, 112, 7707-7712 (2015)
17) Lefèvre C, et al.: Mutations in a new cytochrome P450 gene in lamellar ichthyosis type 3. Hum Mol Genet, 15, 767-776 (2006)
18) Oji V, et al.: Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J Am Acad Dermatol, 63, 607-641 (2010)
19) Traupe H, et al.: Nonsyndromic types of ichthyoses – an update. J Dtsch Dermatol Ges, 12, 109-121 (2014)
20) Schulze H, et al.: Sphingolipids and lysosomal pathologies. Biochim Biophys Acta, 1841, 799-810 (2014)
21) Ehlert K, et al.: Farber disease: clinical presentation, pathogenesis and a new approach to treatment. Pediatr Rheumatol Online J, 5, 15 (2007)
22) Coant N, et al.: Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv Biol Regul, 63, 122-131 (2017)
23) Wakashima T, et al.: Dual functions of the trans-2-enoyl-CoA reductase TER in the sphingosine 1-phosphate metabolic pathway and in fatty acid elongation. J Biol Chem, 289, 24736-24748 (2014)
24) Stoffel W, et al.: Metabolism of sphingosine bases, II. Studies on the degradation and transformation of [3-14C]erythro-DL-dihydrosphingosine,[7-3H]erythro-DL-sphingosine,[5-3H]threo-L-dihydrosphingosine and [3-14C;1-3H]erythro-DL-dihydrosphingosine in rat liver. Hoppe Seylers Z Physiol Chem, 348, 1345-1351 (1967)
25) Kihara A: Sphingosine 1-phosphate is a key metabolite linking sphingolipids to glycerophospholipids. Biochim Biophys Acta, 1841, 766-772 (2014)
26) Nakahara K, et al.: The Sjögren-Larsson syndrome gene encodes a hexadecenal dehydrogenase of the sphingosine 1-phosphate degradation pathway. Mol Cell, 46, 461-471 (2012)
27) Jojima K, et al.: Metabolism of sphingadiene and characterization of the sphingadiene-producing enzyme FADS3. Biochim Biophys Acta Mol Cell Biol Lipids, 1868, 159335 (2023)
28) Kohama T, et al.: Molecular cloning and functional characterization of murine sphingosine kinase. J Biol Chem, 273, 23722-23728 (1998)
29) Liu H, et al.: Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem, 275, 19513-19520 (2000)
30) Kihara A, et al.: Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine 1-phosphate and ceramide 1-phosphate. Prog Lipid Res, 46, 126-144 (2007)
31) Kihara A, et al.: Production and release of sphingosine 1-phosphate and the phosphorylated form of the immunomodulator FTY720. Biochim Biophys Acta, 1781, 496-502 (2008)
32) Zhou J, et al.: Identification of the first mammalian sphingosine phosphate lyase gene and its functional expression in yeast. Biochem Biophys Res Commun, 242, 502-507 (1998)
33) Mandala SM, et al.: Molecular cloning and characterization of a lipid phosphohydrolase that degrades sphingosine-1-phosphate and induces cell death. Proc Natl Acad Sci USA, 97, 7859-7864 (2000)
34) Ogawa C, et al.: Identification and characterization of a novel human sphingosine-1-phosphate phosphohydrolase, hSPP2. J Biol Chem, 278, 1268-1272 (2003)
35) Ikeda M, et al.: Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem Biophys Res Commun, 325, 338-343 (2004)
36) Stoffel W, et al.: Metabolism of sphingosine bases. IX. Degradation in vitro of dihydrospingosine and dihydrospingosine phosphate to palmitaldehyde and ethanolamine phosphate. Hoppe Seylers Z Physiol Chem, 349, 1745-1748 (1968)
37) Stoffel W, et al.: Metabolism of sphingosine bases. X. Degradation of [1-14C]dihydrosphingosine (sphinganine), [1-14C]2-amino-1,3-dihydroxyheptane and [1-14C]dihydrosphingosine phosphate in rat liver. Hoppe Seylers Z Physiol Chem, 350, 63-68 (1969)
38) Bektas M, et al.: Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J Biol Chem, 285, 10880-10889 (2010)
39) Vogel P, et al.: Incomplete inhibition of sphingosine 1-phosphate lyase modulates immune system function yet prevents early lethality and non-lymphoid lesions. PLoS One, 4, e4112 (2009)
40) Janecke AR, et al.: Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications. Hum Mutat, 38, 365-372 (2017)
41) Lovric S, et al.: Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J Clin Invest, 127, 912-928 (2017)
42) Prasad R, et al.: Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J Clin Invest, 127, 942-953 (2017)
43) Marchitti SA, et al.: Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol, 4, 697-720 (2008)
44) Jackson B, et al.: Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum Genomics, 5, 283-303 (2011)
45) Kitamura T, et al.: Substrate specificity, plasma membrane localization, and lipid modification of the aldehyde dehydrogenase ALDH3B1. Biochim Biophys Acta, 1831, 1395-1401 (2013)
46) Kitamura T, et al.: Mouse aldehyde dehydrogenase ALDH3B2 is localized to lipid droplets via two C-terminal tryptophan residues and lipid modification. Biochem J, 465, 79-87 (2015)
47) Ashibe B, et al.: Dual subcellular localization in the endoplasmic reticulum and peroxisomes and a vital role in protecting against oxidative stress of fatty aldehyde dehydrogenase are achieved by alternative splicing. J Biol Chem, 282, 20763-20773 (2007)
48) Naganuma T, et al.: Disruption of the Sjögren-Larsson syndrome gene Aldh3a2 in mice increases keratinocyte growth and retards skin barrier recovery. J Biol Chem, 291, 11676-11688 (2016)
49) Rizzo WB: Sjögren-Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab, 90, 1-9 (2007)
50) Rizzo WB: Fatty aldehyde and fatty alcohol metabolism: review and importance for epidermal structure and function. Biochim Biophys Acta, 1841, 377-389 (2014)
51) Kanetake T, et al.: Neural symptoms in a gene knockout mouse model of Sjögren-Larsson syndrome are associated with a decrease in 2-hydroxygalactosylceramide. FASEB J, 33, 928-941 (2018)
52) Schumacher F, et al.: The sphingosine 1-phosphate breakdown product, (2E)-hexadecenal, forms protein adducts and glutathione conjugates in vitro. J Lipid Res, 58, 1648-1660 (2017)
53) Nojiri K, et al.: Impaired skin barrier function due to reduced ω-O-acylceramide levels in a mouse model of Sjögren-Larsson syndrome. Mol Cell Biol, 41, e0035221 (2021)
54) Arai A, et al.: Ceramide profiling of stratum corneum in Sjögren-Larsson syndrome. J Dermatol Sci, 107, 114-122 (2022)
55) Watkins PA, et al.: Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J Lipid Res, 48 (12), 2736-2750 (2007)
56) Ohkuni A, et al.: Identification of acyl-CoA synthetases involved in the mammalian sphingosine 1-phosphate metabolic pathway. Biochem Biophys Res Commun, 442, 195-201 (2013)
57) Moon YA, et al.: Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J. Biol. Chem., 276, 45358-45366 (2001)
58) Rantakari P, et al.: Hydroxysteroid (17β) dehydrogenase 12 is essential for mouse organogenesis and embryonic survival. Endocrinology, 151, 1893-1901 (2010)
59) Çalişkan M, et al.: Exome sequencing reveals a novel mutation for autosomal recessive non-syndromic mental retardation in the TECR gene on chromosome 19p13. Hum Mol Genet, 20, 1285-1289 (2011)
60) Kitamura T, et al.: Phytosphingosine degradation pathway includes fatty acid α-oxidation reactions in the endoplasmic reticulum. Proc Natl Acad Sci USA, 114, E2616-E2623 (2017)
61) Foulon V, et al.: Breakdown of 2-hydroxylated straight chain fatty acids via peroxisomal 2-hydroxyphytanoyl-CoA lyase: a revised pathway for the α-oxidation of straight chain fatty acids. J Biol Chem, 280, 9802-9812 (2005)
62) Cracan V, et al.: Novel B12-dependent acyl-CoA mutases and their biotechnological potential. Biochemistry, 51, 6039-6046 (2012)
63) Wongkittichote P, et al.: Propionyl-CoA carboxylase – a review. Mol Genet Metab, 122, 145-152 (2017)
64) Tove SB: Production of odd-numbered carbon fatty acids from propionate by mice. Nature, 184, 1647-1648 (1959)
65) Hajra AK, et al.: Biosynthesis of the cerebroside odd-numbered fatty acids. J Lipid Res, 3, 327-332 (1962)
66) Hajra AK, et al.: Isotopic studies of the biosynthesis of the cerebroside fatty acids in rats. J Lipid Res, 4, 270-278 (1963)
67) Svennerholm L, et al.: Changes in the fatty acid composition of cerebrosides and sulfatides of human nervous tissue with age. J Lipid Res, 9, 215-225 (1968)
68) Farwanah H, et al.: Profiling of human stratum corneum ceramides by means of normal phase LC/APCI-MS. Anal Bioanal Chem, 383, 632-637 (2005)
69) Mori K, et al.: Role of 2-hydroxy acyl-CoA lyase HACL2 in odd-chain fatty acid production via α-oxidation in vivo. Mol Biol Cell, 34, ar85 (2023)